免费的Winpwmat GUI版本上线

去年的8月,我们发布了Winpwmat简易软件包版本,目的是为了让更多初学者可以在自己的电脑上进行一些简单的计算,通过这个方式让新手能够更快得学习、熟悉PWmat的功能,或者从简单基础开始产生对DFT计算的兴趣,做到“材料计算、随时可算”。
受到广大用户热情的“催更”,这次,我们正式发布升级后的Winpwmat GUI版本。完全免费软件, 特别为教学、学生学习而开发。在原有基础上GUI版实现安装后电脑桌面图标展示、产品图形界面等功能,方便新用户上手熟悉PWmat的基础功能,同时还可以在界面中调用另一款产品Q-Studio,掌握成熟的建模能力,可以自己搭建结构或从数据库下载结构文件,实现了一整套无障碍计算流程。
Winpwmat GUI版本大大降低了计算的门槛,无需操作不熟悉的powershell命令行以及耗费学习成本来准备输入文件,实现了一键式的计算,即使是小白用户也可以依靠它快速获取计算结果。
Winpwmat GUI版本
Winpwmat目前可用的功能包括结构优化(JOB=RELAX)、能量计算(自洽计算JOB=SCF)、分子动力学模拟(JOB=MD)、BAND(能带)计算以及DOS(态密度)计算。拥有一台笔记本电脑即可开始,真正做到零门槛做计算,即使是刚刚接触该类学科的小白,也可以通过界面实现用自己的电脑做一些小体系计算,并对结果做一些便捷的可视化分析。
使用中提供两套计算方案
快速开始
默认参数

无需参数设置的“快速开始”可以进行精度较低的默认参数计算,一键实现计算
为进阶用户提供的详细参数设置,得到更高精度的计算结果
高级计算
详细参数设置
Winpwmat使用说明书
01
设定工作路径
点击左上方“快速开始”→“STEP1:设置本项目文件夹”,选择一个本地工作路径

02
创建你的结构(Q-Studio)
若已有结构文件可通过“快速开始”→“STEP2:构建结构”→“本地上传”,将已有的atom.config传至当前文件夹

若没有结构,通过点击“快速开始”→“STEP2:构建结构”→“通过Q-Studio构建

打开Q-Studio来构建结构,结合Q-Studio教程↓
https://space.bilibili.com/1965576887/channel/collectiondetail?sid=2114809
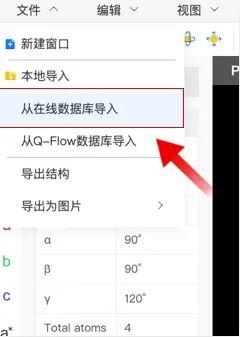
点击“文件”→“新建窗口”新建一个结构,或“从在线数据库导入”一个结构后,点击“导出结构”可以保存至当前文件夹下


提示: 计算将会以atom.config作为结构文件进行计算,若是从Q-Studio上下载的以PWmat为尾缀的结构文件在运行时将被自动转换为atom.config
03
设置工作MPI并行数
在开始之前可以通过“快速开始”→“STEP3:设置工作MPI并行数”来设置并行的数量,默认为1

04
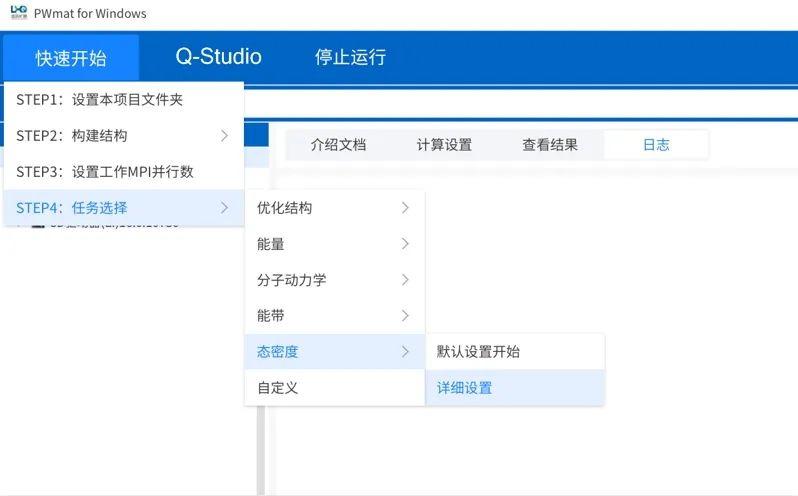
任务选择
点击菜单“快速开始”→“STEP4:任务选择”可以选择进行的计算任务,点击“默认设置开始”,即可进行计算。
提示: 计算将会以atom.config作为结构文件进行计算,若是以PWmat为尾缀的结构文件在运行时将被自动转换为atom.config
05
其他提示
若在当前文件夹下找不到结构文件会在右上角提示

如提示所说,你可以点击“提示”上传一个结构文件至本地,或点击“Q-Studio”创建一个结构

06
以态密度为例继续计算
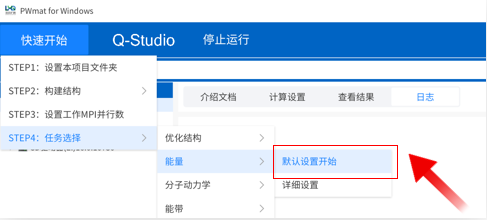
默认参数快速开始
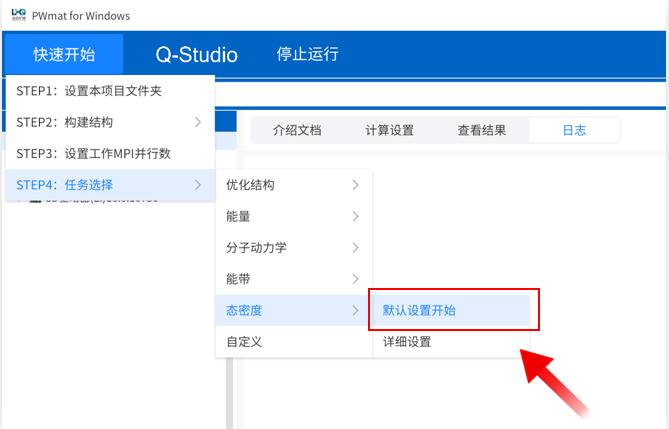
点击菜单“快速开始”→“STEP4:任务选择”→“态密度”→“默认设置开始”,下方进度条显示计算进度
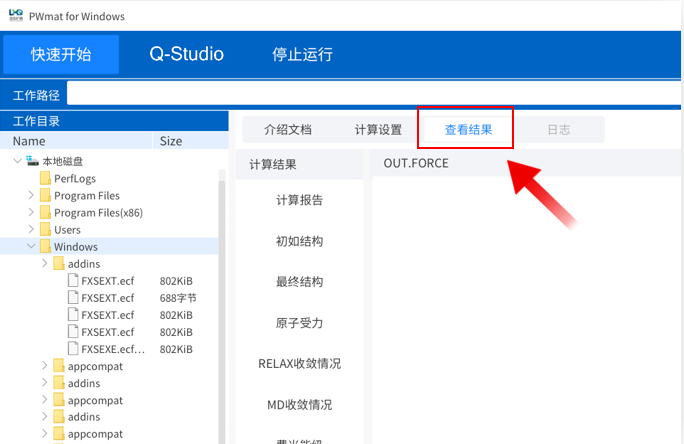
计算成功后点击“提示”跳转结果分析,或者点击菜单“查看结果”

详细参数设置高级计算
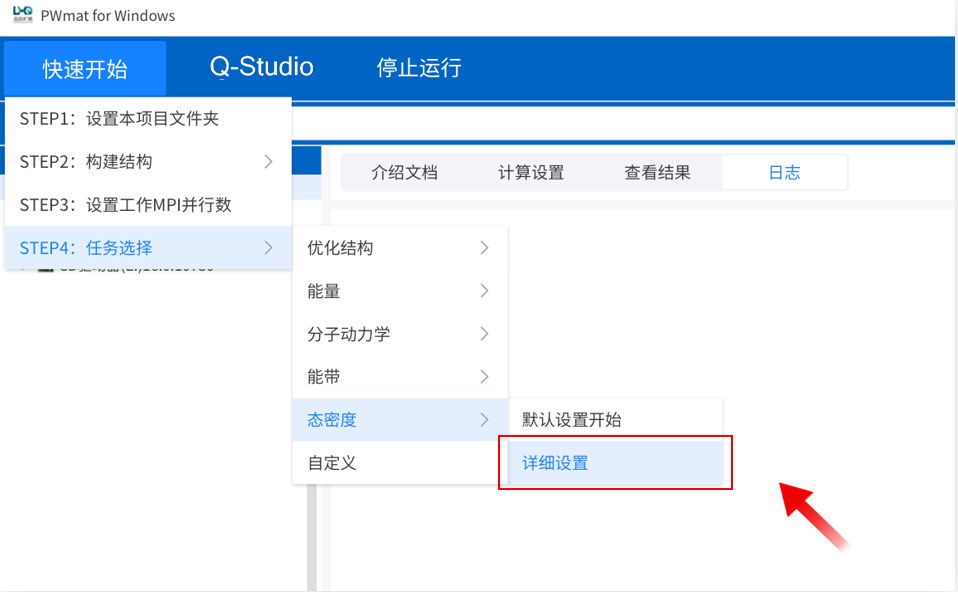
点击菜单“快速开始”→“STEP4:任务选择”→“态密度”→“详细设置”
点击“STEP:SCF”设置第一步SCF计算的etot.input,修改完成后点击保存更改;
点击“STEP:DOS”设置第二步态密度的etot.input,修改完成后点击保存更改

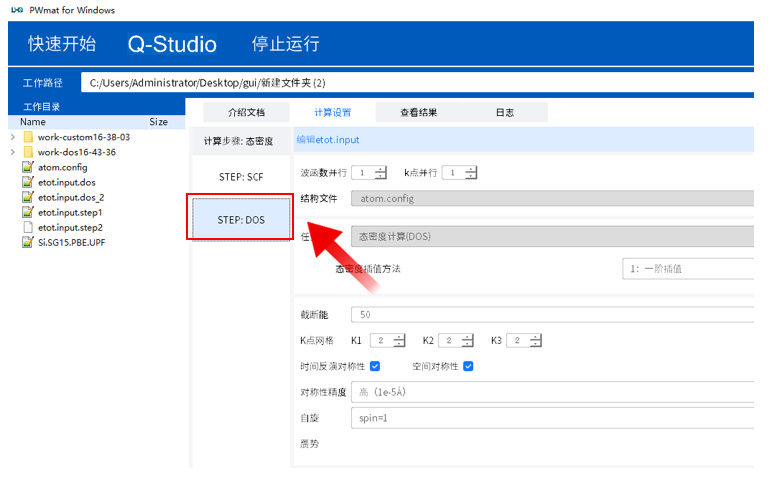

在设置完成后点击“保存更改”会在本地产生相应的输入文件。两步都保存之后,点击菜单中的“运行”即可开始计算
07
查看结果
进入“查看结果”界面,点击计算结果内的“原子受力”,即可分析计算得到的原子受力情况

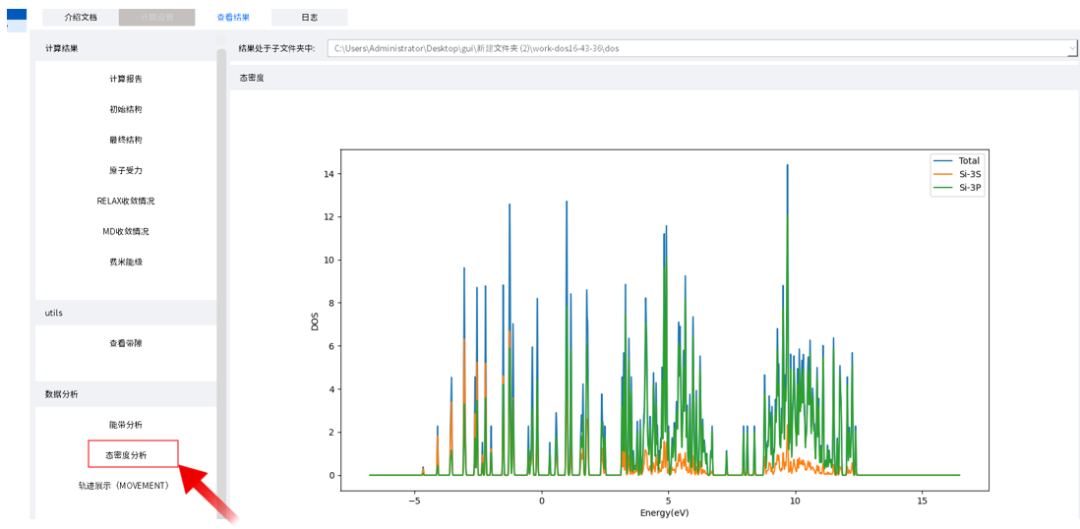
以刚刚的态密度为例,想要查看态密度计算结果需要先切换至结果存在的子文件夹dos中

在切换至子文件夹后,点击左侧下方“态密度分析”,即可查看态密度,并且可以在图像下方修改横坐标