ORR计算案例说明,复刻《Journal of Physical Chemistry》学术文献

环境污染和能源短缺是21世纪以来人类社会面临的两大难题,因此寻找清洁可再生能源以及开发高效的能量存储和转换技术是当务之急。催化反应是多种能量转换器件的核心过程,而催化研究的核心就是研究四大反应(HER、OER、ORR、HOR),例如水分解反应就可以分为析氢反应(HER)和析氧反应(OER)两个半反应进行研究,而对于氧化还原反应(ORR)是能量转化反应中非常重要的一环,在燃料电池以及锂空电池等方面都用重要的应用。通过四大反应阐述清楚催化机理,对我们无论是从理论计算还是实验方面设计更好的电催化剂都是非常必要的。在研究过程中,很多初学者苦于不熟悉做一个具体流程,而最好的学习方法就是跟着经典例子摸索一遍,这将增加学习效率,更快得补齐知识短板,发现新的研究方向。
“
本教程以石墨烯单铁原子缺陷结构 Fe/SVG 为例,主要展示其催化氧还原反应路径的自由能计算步骤,其中包括建模过程、PWmat软件参数设置、计算结果处理等。文中使用的是2021-11-02 的PWmat版本。
1、石墨烯单胞优化
1.1 石墨烯建模
为了得到 Fe/SVG,我们需要先进行石墨烯优化。对于大部分二维材料,真空层取15Å左右便已足够。
1.2 准备输入文件
a. 结构文件
准备好石墨烯的 graphene.cif 文件,上传至计算目录,通过 cif2cell 命令将cif 格式转换为 PWmat 格式:
cif2cell graphene.cif -p pwmat
运行成功会生成 atom.config 文件,即为PWmat 格式的结构文件,二维材料优化需要固定 z 方向晶格,在atom.config 中加入STRESS_MASK 选项:

b. 输入参数文件
需要同时优化晶格、原子位置,etot.input 如下:
1 4
JOB = relax
IN.ATOM = atom.config
IN.PSP1 = C.SG15.PBE.UPF
RELAX_DETAIL = 1 100 0.01 1 0.01
ECUT = 70
ECUT2 = 280
MP_N123 = 15 15 1 0 0 0
c. 赝势文件与 pbs 脚本文件
略。
1.3 计算与结果处理
提交任务进行计算,优化结束后,将 final.config 文件保存为graphene_unitcell.config文件。
2、Fe/SVG 优化
2.1 Fe/SVG 建模
通过 atomconfig2cif、cif2cell 命令将 graphene_unitcell.config 转换为4×4×1 的超胞:
atomconfig2cif graphene_unitcell.config
cif2cell graphene_unitcell.config.cif -p pwmat --supercell=[4,4,1]
运行成功会生成 atom.config 文件,即为 PWmat 格式的石墨烯超胞结构文件。
将 atom.config 文件中的某一个 C 原子修改为 Fe 原子,并修改Fe 原子z 方向位置,使其略高于石墨烯表面,具体过程略。

2.2 准备输入文件
a. etot.input
只需要优化原子位置,但需要开启自旋极化计算和 VDW 修正,etot.input 如下:
4 1
JOB = relax
IN.ATOM = atom.config
IN.PSP1 = C.SG15.PBE.UPF
IN.PSP2 = Fe.SG15.PBE.UPF
RELAX_DETAIL = 1 100 0.01
ECUT = 60
ECUT2 = 240
MP_N123 = 3 3 1 0 0 0
SPIN = 2
VDW = DFT-D3
b. 结构文件
需要在 atom.config 文件中设置初始磁矩,具体过程略。
c. 赝势文件与 pbs 脚本文件
略。
2.3 计算与结果处理
提交任务进行计算,优化结束后,final.config 即为最终的 Fe/SVG 结构。

完成初始建模工作后,我们可以开始正式的自由能计算步骤!

3、溶剂效应下修正的体系基态能量计算
3.1 准备输入文件
a. etot.input
需要开启溶剂效应模型,etot.input 如下:
4 1
JOB = SCF
IN.ATOM = atom.config
IN.PSP1 = C.SG15.PBE.UPF
IN.PSP2 = Fe.SG15.PBE.UPF
RELAX_DETAIL = 1 100 0.01
ECUT = 60
ECUT2 = 240
MP_N123 = 3 3 1 0 0 0
SPIN = 2
VDW = DFT-D3
CONVERGENCE = DIFFICULT
IN.SOLVENT = T
b. 溶剂模型输入文件
模拟水溶液环境,IN.SOLVENT 文件如下:
DIELECTRIC_CONST = 78
SURFACE_TENSION = 50
PRESSURE = -0.35
RHOMAX_DIELECTRIC = 0.005
RHOMIN_DIELECTRIC = 0.0001
DIELECTRIC_MODEL = ATOM_CHARGE
PARAM_CHARGE.1 = 1.0, 1.0, 1.00
PARAM_CHARGE.2 = 1.0, 1.0, 1.00
RHOMAX_CAVITY = 0.005
RHOMIN_CAVITY = 0.0001
c. 赝势文件与 pbs 脚本文件
略。
3.2 计算与结果处理
得到修正后的 Fe/SVG 的总能 E
4、HOO-Fe/SVG、(HO)2-Fe/SVG、O-Fe/SVG、HO-Fe/SVG 的结构优化及其在溶剂效应下修正的总能计算请参考步骤 2、3,该 4 种结构模型如下图:

5、零点能 ZPE 计算
首先安装 PyPWmat 模块
对于吸附结构,仅计算吸附体的声子振动频率即可,H2及 H2O 分子则可以采用直接计算的形式,这里以吸附结构声子频率计算为例。
5.1 准备输入文件
a. 结构文件
拷贝 atom.config 为 atom_all.config,修改除吸附原子之外的原子的后三列为0:

b. 参数文件
拷贝 etot.input 为 etot_sub.input,修改后如下:
4 1
JOB = scf
IN.ATOM = atom.config
IN.PSP1 = C.SG15.PBE.UPF
IN.PSP2 = Fe.SG15.PBE.UPF
IN.PSP3 = O.SG15.PBE.UPF
ECUT = 60
ECUT2 = 240
MP_N123 = 3 3 1 0 0 0
SPIN = 2
VDW = DFT-D3
CONVERGENCE = DIFFICULT
OUT.FORCE = T
c. 赝势文件与 pbs 脚本文件
略。
d. 声子计算文件
运行 PWmatPhonon.py,产生 PWphonon.in 文件,修改 JOB 参数为sub;DIM、MP
参数为 1 1 1。
5.2 计算与结果处理
再次运行 PWmatPhonon.py 进行声子计算,声子计算详细教程可参见官网。从mesh.yaml.dat 中读取声子振动频率,根据公式:

计算零点能。需要注意的是对于 H2(线性分子)和 H2O(非线性分子)需分别去掉最低的 5 个和 6 个频率。
6、写出 O2经过四电子途径生成 H2O 的 2 种反应途径,区别在于第二步是生成∗O还是∗(OH)2:

根据上述公式可得:
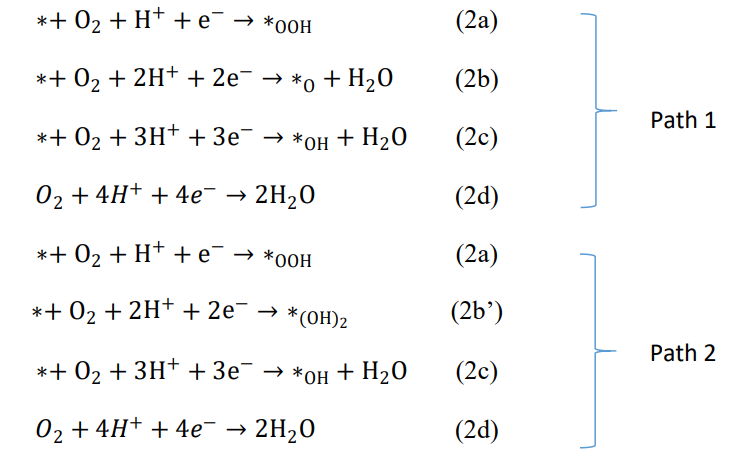
由以上公式可得每个台阶的∆G:

其中μe为电子自由能。在 PH=0 时,有公式 G(H+) + μe(SHE) = 0.5G(H2),其中μe(SHE)为标准氢电极,因此我们可以定义μe = μe(SHE) − U,其中 U 为以标准氢电极为参考点的外加电势,带入上述各方程可得:

其中 G(O2) = 2G(H2O) − 2G(H2) + 4 ∗ 1.23eV。根据自由能计算公式G = E +ZPE −TS,其中 E 为溶剂模型修正的各体系总能,ZPE 为零点能,TS 项为声子振动熵,对于吸附结构来说,TS 项可以忽略,对于 H2和 H2O,TS 项可以通过查找 CSC 手册或参考文献得到。
统计频率并计算 ZPE 及自由能,参考如下(部分):

带入统计好的数据可得:
∆G1 =− 1.275 eV
∆G2 =− 2.07 eV
∆G2' =− 2.27 eV
∆G3 =− 1.99 eV
∆G4 = 0
可得反应路径与自由能变化关系图如下:

“
本教程案例来源
Yan, M., Dai, Z., Chen, S., Dong, L., Zhang, X. L., Xu, Y., & Sun, C. (2020). Single-ironsupported on defective graphene as efficient catalysts for oxygen reduction reaction. The Journal of Physical Chemistry C, 124(24), 13283-13290.


