一条通往开源GROMACS分子动力学模拟仿真捷径(附视频教程)
- 作者优秀
- 优秀教师/意见领袖/博士学历/特邀专家/独家讲师
- 平台推荐
- 内容稀缺




本教程内容适用于Gromacs,包括:
3. 两相共存状态下水结冰过程的模拟;
TIP4P是一种四点水模型,亦即除了一个O和2个H原子外还加入了一个虚原子(在氧附近),虚原子仅具有负电荷,改善了水分子周围的静电分布
其中,TIP4P/ICE是专门用来描述冰的一系列性质的水模型,对水的相图有较好的还原(1bar下冰-Ih熔点为272.2K)。而Gromacs的力场中没有自带该水模型,需要到gromacs官网获取(内容如下)。
;; Note the strange order of atoms to make it faster in gromacs.;[]; molnamenrexclSOL2[]; idat typeres nr residu nameat namecg nrcharge1 O 1 SOL O 1 0.002 H 1 SOL H 1 0.58973 H 1 SOL H 1 0.58974 M 1 SOL M 1 -1.1794[];i j funct doh dhh1 2 1 0.095721 3 1 0.095722 3 1 0.15139[]1234213431244123; The position of the virtual site is computed as follows:;;O;; D;;HH;; const = distance (OD) / [ cos (angle(DOH)) * distance (OH) ]; 0.01577 nm/ [ cos (52.26 deg)* 0.09572 nm]; Vsite pos x4 = x1 + a*(x2-x1) + b*(x3-x1)[]; Vsite fromfunctab412310.13458335 0.13458335
来使用新的水模型。
注意你需要根据使用的力场来修改以上文件中的at type,如对于OPLS-AA,原子类型修改为
[ atoms ]; idat typeres nr residu nameat namecg nrcharge1 opls_113 1 SOL O 1 0.002 opls_114 1 SOL H 1 0.58973 opls_114 1 SOL H 1 0.58974 opls_115 1 SOL M 1 -1.1794
总之就是要修改为这个力场中有定义且定义正确的原子类型。你可以参照力场文件夹中其它水模型使用的原子类型来修改。
当然,你也可以直接简单粗暴地将以上水的拓扑文件直接放在当前模拟的拓扑文件中。没有适当模板的结晶模拟是非常困难的,因此在本模拟中使用冰-Ih/水两相体系。使用packmol建模,输入代码如下。上下填充两层冰,中间充满水
tolerance 2.0output icebox.pdbfiletype pdbstructure ice.pdbnumber 1inside box 0. 0. 0. 28. 24. 23.end structurestructure ice.pdbnumber 1inside box 0. 0. 46. 28. 23. 69.end structurestructure water.pdbnumber 600inside box 0. 0. 23. 28. 23. 46.end structure
得到的icebox.pdb若要直接用于模拟,还需要编辑模拟盒子信息,在最后一行REMARK后加入以下信息
CRYST1 28 23 69 90.00 90.00 90.00 P 1 1该信息记载了模拟盒子的晶胞参数(三边长,角度等),可被gromacs读取。
至此我们得到了三点水模型的坐标文件,先在spc/e模型下进行能量最小化
gmx grompp -f em.mdp -c icebox.pdb -p icebox.top -o em.tpr -maxwarn 2gmx mdrun -v -deffnm em
这样就得到了经过能量最小化的em.gro。接着,我们要在不改变原坐标的情况下,给所有水引入虚原子,转化为四点水模型的坐标文件。运行
cat em.gro |awk '{print $0;if($2=="OW"){a=$1;b=$3;c=$4;d=$5;e=$6;}if($2=="HW2")printf("%8s MW %4d%8.3f%8.3f%8.3f\n",a,b,c,d,e)}' > newicebox.gro这段代码意思是逐行读取em.gro文件,读到氧原子一行时,将氧原子的编号和坐标写入临时变量abcde,然后读到每个水分子的第二个氢原子时在下一行插入虚原子,编号和坐标与第一个氧原子相同。亦即在每个水的氧原子位置添加一个虚原子。
然后在修改过的使用tip4p/ice水模型的拓扑文件下重新进行能量最小化(由于新引入了虚原子,你需要手动修改gro文件的原子数量)
gmx grompp -f em.mdp -c newicebox.gro -p icebox4.top -o em.tpr -maxwarn 2gmx mdrun -v -deffnm em
由于之前已经在三点水下进行过能量最小化,所以这一步能量最小化只需要调整虚原子位置即可,一步优化就完成了。
这样就得到了基于TIP4P的冰/水坐标文件。
模拟照常在能量最小化之后再进行NVT、NPT预平衡,然后进行产生相模拟。这里贴出产生相模拟所使用的mdp文件,其它mdp文件可在开头链接中的附件获取。
; Run parametersintegrator = md ; leap-frog integratornsteps = 100000000 ; 1 * 100000000 = 10000 ps (10 ns)dt = 0.001 ; 1 fs; Output controlnstxout = 4000 ; save coordinates every 0.4 psnstvout = 4000 ; save velocities every 0.4 psnstenergy = 4000 ; save energies every 0.4 psnstlog = 4000 ; update log file every 0.4 psnstxtcout = 40000 ; xtc compressed trajectory output every 40 ps; Bond parameterscontinuation = yes ; Restarting after NVTconstraint_algorithm = lincs ; holonomic constraintsconstraints = h-bonds ; only h-bonds constrainedlincs_iter = 1 ; accuracy of LINCSlincs_order = 4 ; also related to accuracy; Neighborsearchingns_type = grid ; search neighboring grid celsnstlist = 5 ; 5 fsrlist = 1.0 ; short-range neighborlist cutoff (in nm)rcoulomb = 1.0 ; short-range electrostatic cutoff (in nm)rvdw = 1.0 ; short-range van der Waals cutoff (in nm); Electrostaticscoulombtype = PME ; Particle Mesh Ewald for long-range electrostaticspme_order = 4 ; cubic interpolationfourierspacing = 0.16 ; grid spacing for FFT; Temperature coupling is ontcoupl = v-rescale ; More accurate thermostattc-grps = ICE SOL ; two coupling groups - more accuratetau_t = 0.5 0.5 ; time constant, in psref_t = 260 260 ; reference temperature, one for each group, in K; Pressure coupling is onpcoupl = Parrinello-Rahman ; Pressure coupling on in NPTpcoupltype = semiisotropic ; uniform scaling of x-y box vectors, independent ztau_p = 5.0 ; time constant, in psref_p = 450.0 450.0 ; reference pressure, x-y, z (in bar)compressibility = 4.5e-5 4.5e-5 ; isothermal compressibility, bar^-1; Periodic boundary conditionspbc = xyz ; 3-D PBC; Dispersion correctionDispCorr = EnerPres ; account for cut-off vdW scheme; Velocity generationgen_vel = no ; Velocity generation is off; COM motion removal; These options remove motion of the ice relative to the solvent/ionsnstcomm = 1comm-mode = Linearcomm-grps = ICE SOL; Scale COM of reference coordinatesrefcoord_scaling = com
在这里,步长dt=1fs, 使用V-rescale控温,参考温度260K, Parrinello-Rahman 控压, 参考压力为 450bar 。
使用了半各向异性控压,并将冰和水分开来控温——不过这也许并不必要。
以上条件显然不算最优的,还需要按照自己需求进行调整参数。我实际模拟了300ns,能够观察到结冰过程。
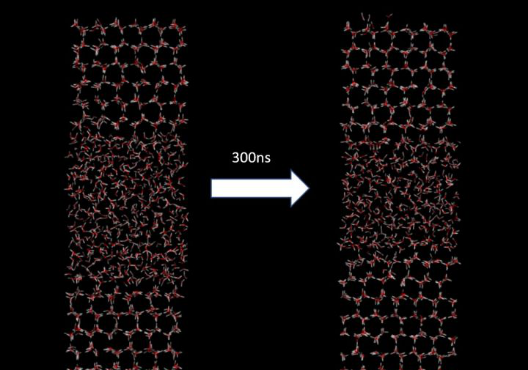
除了直观地观察冰晶的形成情况之外,还可以通过统计氢键平均寿命来衡量结晶的程度。
三、我的GROMACS热门教程
总之,GROMACS是用于研究生物、化学、物理等学科中分子微观机理的分子动力学程序包。它可以用分子动力学、随机动力学或者路径积分方法模拟溶液或晶体中的任意分子,进行分子能量的最小化,分析构象等。
它的模拟程序包含GROMACS力场、AMBER力场等诸多力场,研究的范围可以包括玻璃和液晶、聚合物、晶体、界面体系和生物分子溶液(蛋白质、核苷酸、糖等)。GROMACS是一个功能强大的分子动力学的模拟软件,其在模拟大量分子系统的牛顿运动方面具有极大的优势。
为此,笔者录制了Gromacs系列视频课程,开通一条通往开源Gromacs分子动力学模拟仿真捷径,现成为网上Gromacs热门课程,受到了数千人的好评。
1、零基础入门进阶教程
GROMACS分子动力学模拟仿真22讲:自学冰热融化、离子溶液、界面模型、纳米管和蛋白建模仿真分析


扫码立即试看
该课程拟在为从事上诉科研领域人员提供GROMACS软件专业培训,提高相关人员的技术水平。
无论您前期是否接触过此软件,均可通过此次培训更加熟练的运用本领域的计算仿真。
该课程通过由易到难的知识讲解,让您对GROMACS软件使用有一个全面系统的学习,更好的补充您在自学中忽略的知识内容。
2、GROMACS专题进阶课程

扫码领取优惠券
专题一、 GROMACS生物膜/膜蛋白的构建和模拟

本专题视频课程内容包括:
(4)膜蛋白的构建和模拟:膜蛋白和DPPC为例
专题二:GROMACS含有金属配件的蛋白建模与模拟

(1)处理金属离子的模拟参数,生成蛋白-金属离子的仿真盒子
(2)金属离子和蛋白没有任何成键作用,不用考虑模拟过程离子是否偏离结合位点的模拟
(3)金属离子和蛋白最近的氨基酸有键连关系的模拟
专题三、GROMACS质心牵引(伞形采样)模拟仿真计算

(1)对目标结构进行预处理
(2)进行质心牵引模拟
专题四、GROMACS非标准残基力场构建和高分子itp构建

本专题视频教程课程目录:
(3)如何生成高分子的ITP/TOP文件
专题五、GROMACS粗粒化力场建立和模拟仿真

本专题视频教程课程目录:
(1)溶液中的蛋白粗粒化模拟
(2)小分子粗粒化构建
(3)磷脂膜、混合膜粗粒化的构建和模拟
(4)膜蛋白粗粒化的构建和模拟
(5)DNA、RNA粗粒化的构建和模拟
专题六、GROMAC TOP-ITP文件使用和注意事项

专题七、GROMACS虚拟墙(基底)的实现

专题八、GROMACS二氧化硅-石墨烯-氧化石墨烯等周期性体系的构建方法

(1)石墨烯-碳纳米管-氧化石墨烯建模
专题九、GROMACS多糖-纤维素结构建立

本次专题讲解如何搭建多糖、纤维素等结构文件以及如何生成GROMACS所需的ITP文件。
(1)多糖结构、力场文件的生成——glycam力场为例
(2)多糖结构、力场文件的生成——Amber力场、OPLS力场为例
专题十、GROMACS胶原纤维构建

专题十一、GROMACS不同PH下蛋白和小分子模拟仿真

本视频课程主要讲解:GROMACS不同PH下蛋白和小分子的模拟仿真。为付费用户提供VIP交流群和知识圈答疑,欢迎订阅用户联系仿真秀官方客服加入VIP群。讲师会根据用户交流和需要酌情加餐录制视频教程和直播等。
(完)


