Energy Environ. Sci:析氢反应过程中的瞬态相变


研究背景
众所周知,材料的性质,包括催化活性都是相敏感的。而材料的相与外界条件有关,电催化反应过程中的外部条件与反应前后的外部条件不同,这自然导致了一个问题:在反应过程中电催化剂的相是否可以与反应前后的相不同?在这项工作中,作者通过研究电化学析氢反应(HER)过程中2H-MoS2的动态变化,肯定地回答了这个问题,对多相电催化剂的设计和多相电催化机理的研究具有重要意义。
文章简介
2H-MoS2是一种很有前景的无贵金属析氢反应(HER)电催化剂。关于其HER催化机制,人们普遍认为其边缘位点具有较高的HER活性,而基面在HER过程中是惰性的。近日,华中科技大学翟天佑教授团队和浙江师范大学杨发教授团队研究表明这种观点是有问题的。该团队通过第一性原理计算以及原位测量,发现2H-MoS2在HER过程中经历从惰性2H相到1T’相的局部相变,1T’相的部分基面表现出高HER活性,并且这种相变是短暂的,在反应结束后恢复到2H相。本工作揭示了一种新的催化反应机理——瞬态相变,为多相电催化剂的设计和机理研究提供了新的机会。相关研究成果发表在了国际著名期刊《Energy & Environmental Science》 上,论文第一作者为华中科技大学材料学院副教授赵英鹤,本科生李浩博(目前在上海交大攻读博士)以及在读博士生杨若欧。本文计算部分采用PWmat完成。
主要内容
本工作首先使用电荷中性法(charge-neutral method, CNM)计算了2H-MoS2表面S原子的ΔG为1.92 eV(如图1(b)所示),表明其对HER极具惰性。该计算结果与前人计算结果吻合良好。从热力学角度来看,2H-MoS2的H原子与表面S原子之间成键需要约-2.0 V电压。然而,Chen等人在研究2H-MoS2催化HER时,在一个相当低的电位(仅-0.2 V)下实验观察到了表面上S–H键的形成。具体来说,其在2532 cm-1处观察到一个峰值,并将这个峰归因于表面上S–H 键的伸缩振动。
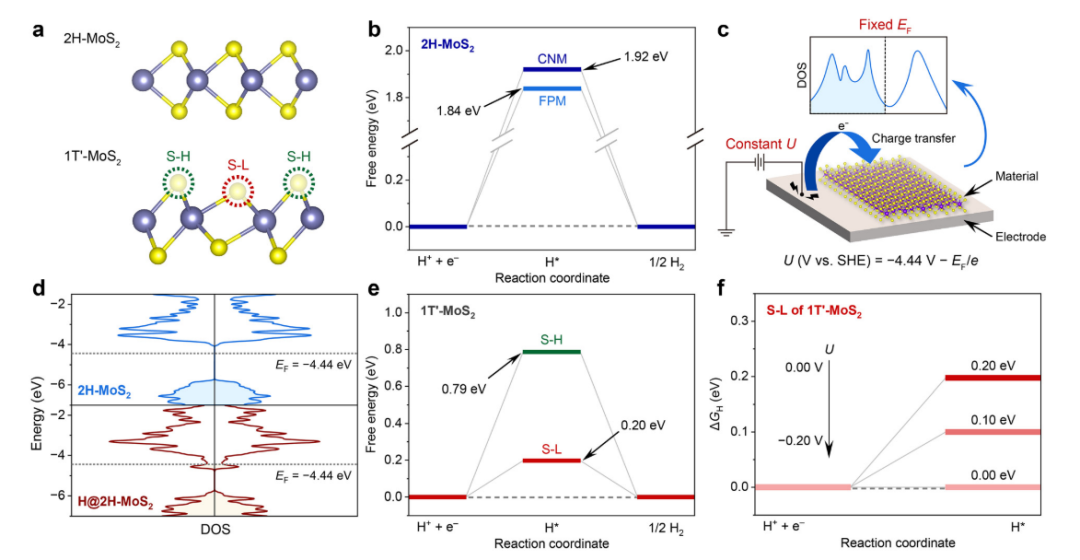
图1 (a) 2H-MoS2和1T’-MoS2侧视图,1T’-MoS2的高、低位置的S原子分别表示为S-H和S-L;(b) U = 0 V时2H-MoS2表面S原子HER过程的自由能图;(c) FPM方法的原理图;(d) U = 0 V时,原始2H-MoS2和2H-MoS2与H吸附原子的态密度,其中EF代表费米能级;(e) U = 0 V时,S-H和S-L的自由能图;(f) S-L的ΔG随电极电位的变化。
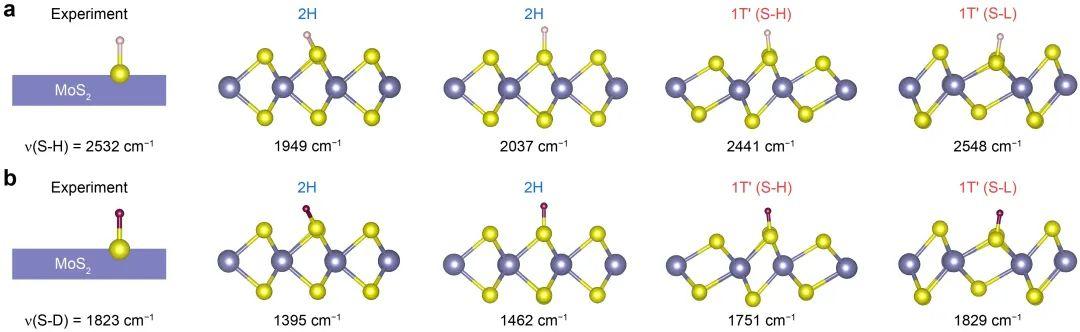
图2 2H-MoS2和1T’-MoS2上υ(S–H)和υ(S–D)的实验测量频率与计算频率的比较。实验值来源于[J. Am. Chem. Soc., 2020, 142, 7161]。
01
排除实验和计算矛盾是由计算方法导致
先前关于2H-MoS2表明氢吸附自由能的计算都是基于固定电荷法获得的,然而由于没有考虑电极电势的影响,固定电荷法被认为是不准确的。所以本工作首先怀疑是否实验和计算的矛盾是由计算方法导致的。相应地作者们,使用固定电势法(fixed-potential method, FPM)计算出了2H-MoS2表面S原子的ΔG,如图1(b)所示,较大的ΔG(1.84 eV)排除了是由于计算方法不同导致与实验结果不符。
02
排除实验和计算矛盾是由S空位导致
考虑到s空位在2H-MoS2中普遍存在,S空位扰动了周围S原子的电荷密度分布(图S7),从而使靠近空位的S原子可能被激发具有HER活性。本工作使用CNM和FPM探究了与S空位附近3考虑到S空位在2H-MoS2中普遍存在,S空位会扰动周围S原子的电荷密度分布,从而使靠近空位的S原子可能被激发具有HER活性。本工作使用CNM和FPM探究了S空位附近3个S原子(如图3(a)所示,分别为S1、S2和S3)的ΔG,计算结果分别如图3(b)和(c)所示。两种计算方法均显示出同样的趋势,即S原子离空位越远,ΔG越大。但是与空位相邻的S原子仍然需要一个高于1.0 V的电位来驱动S–H键的形成。因此,S空位的扰动并不是理论和实验观点产生矛盾的根本原因。个S原子(如图3(a)所示,分别为S1、S2和S3)的ΔG,计算结果分别如图3(b)和(c)所示。两种计算方法均显示出同样的趋势,即S原子离空位越远,ΔG越大。但是与空位相邻的S原子仍然需要一个高于1.0 V的电位来驱动S- H键的形成。因此,S空位的扰动并不是理论和实验观点产生矛盾的根本原因。
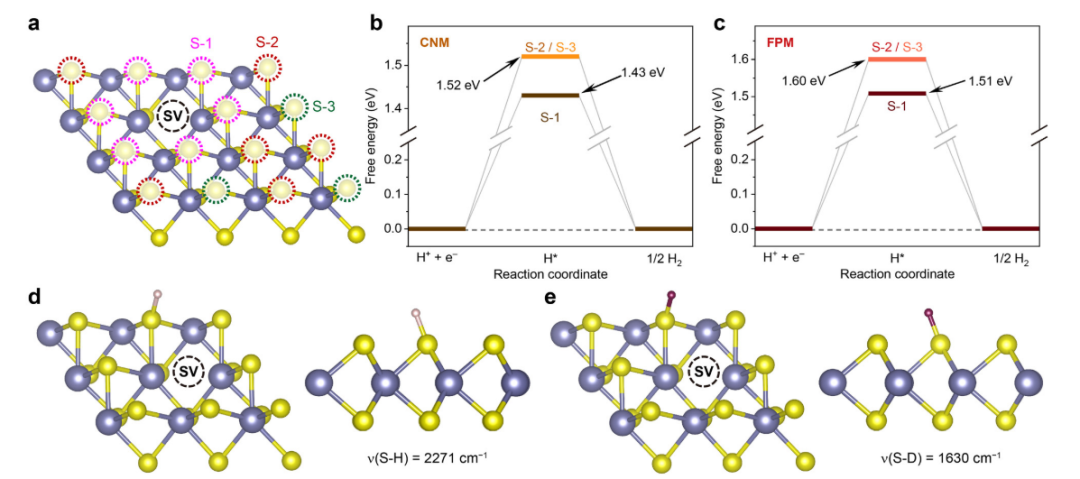
图3 (a) 含S空位的2H-MoS2的结构;(b,c) U = 0 V时S空位附近S原子的HER自由能图;(d,e) 2H-MoS2的S空位附近S原子的υ(S–H)和υ(S–D)频率的理论值。
03
确定实验和计算矛盾是由相变导致
考虑到过渡金属二卤族化合物有两种典型的相:H相和T相(如图1(a)所示),本工作探究了不同相结构下2H-MoS2的HER活性。其中,T相具有两种结构,1T相和1T’相(也称为畸变1T相)。由于1T相吸附H原子会变为1T’相,所以本工作选择1T’相结构来进行研究。由图1(a)可知,1T’-MoS2的结构畸变产生了两种类型的S原子:S-H(S原子位于较高位置)和S-L(S原子位于较低位置)。因此,对两种不同类型的表面S原子进行了计算,结果如图1(e)所示,S-H的ΔG较大(0.79 eV),而S-L的ΔG较小(0.20 eV)。当U= -0.2 V时,ΔG降至0 eV(图1(f)),表明-0.2 V的电位可以促进S-L上S-H键的形成。有趣的是,代表S-H键的信号出现的起始电位也是-0.2 V。二者的一致性表明,MoS2的2H 到1T’相变可以解释实验和计算上存在的矛盾。
由于检测到的S–H键伸缩振动(υ(S–H))是支持表面S–H键形成的直接实验证据。本工作计算了拉伸振动υ(S-H),并将理论结果与实验结果进行了比较(图2)。对于2H-MoS2来说,S–H键在表面的两种吸附模式的υ(S–H)的计算值(1949和2037 cm-1)均与实验值存在较大偏差(图2)。对于1T’-MoS2,在S-H和S-L上的υ(S–H) 计算值分别为2441和2548 cm-1。其中,S-L上的H吸附的υ(S–H)计算值与实验值吻合得很好,再一次表明HER过程中2H-1T’相变可以很好地解决理论与实验之间明显的矛盾。当把H原子换成D原子后,结论不变(图2)。再者,本工作还计算了邻近S空位的S原子上的υ(S–H)和υ(S–D)以及1.5%拉伸应变和1.5%压缩应变下2H-MoS2上υ(S–H)和υ(S–D)的频率,计算值与实验结果均有较大偏差,进一步说明S空位和实际情况中可能存在的微小应变都不是引起矛盾的根本原因。
最后,原位XAFS结果证实了部分2H-MoS2在HER过程中确实可以转变为1T’-MoS2。具体地说,原位XAFS结果表明在初始状态下并没有检测到1T’相的成分,但是当电压达到-0.2V时候显现出了1T’相的特征,并且在反应后1T’相的特征会消失(图4)。
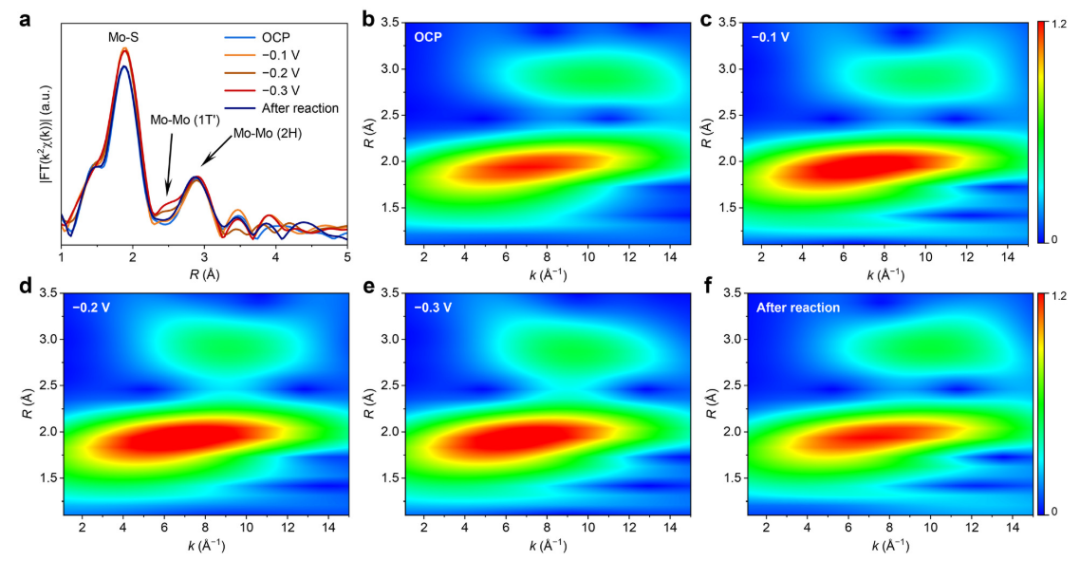
图4 (a) 2H-MoS2在OCP、-0.1 V、-0.2 V、-0.3 V和可逆氢电极(RHE)以及反应后的原位EXAFS光谱;(b-f) 2H-MoS2在(b) OCP、(c) -0.1 V、(d) -0.2 V、(e) -0.3 V和(f)反应后的WT分析。
综上所述,1T’-MoS2表面形成S–H键的起始电位,S-L上的υ(S–H)和υ(S–D)值以及XAFS原位测量均可以证明,将矛盾的原因与相变联系起来是相当合理的。由于S-L的ΔG较小,1T’-MoS2的基面具有较高的HER活性,因此,由于其瞬态相变行为,2H-MoS2的部分基面可以表现出较高的HER活性。本工作已经提供了足够的证据支持2H-MoS2在HER过程中的相变,但相变的驱动机制仍是一个未解之谜,需要通过更多的研究来进一步了解相变的机理。
—E N D—


