药监局已开始对医疗器械电磁兼容进行产品抽样检验 EMC问题日益彰显

日前,北京市药监局发布了医疗器械质量公告。
3月23日,北京市药监局发布了《2016年下半年北京市医疗器械质量安全公告》。
2016年下半年,全市完成对医疗器械生产、经营和使用单位的监督抽样362批次,完成检验592批次,不合格产品25批次。

北京某医疗器械公司生产的强脉冲光治疗仪在抽样检验的时候,产品的电磁兼容性没有符合规定要求,其中传导发射项和辐射发射项存在测试不合格问题。

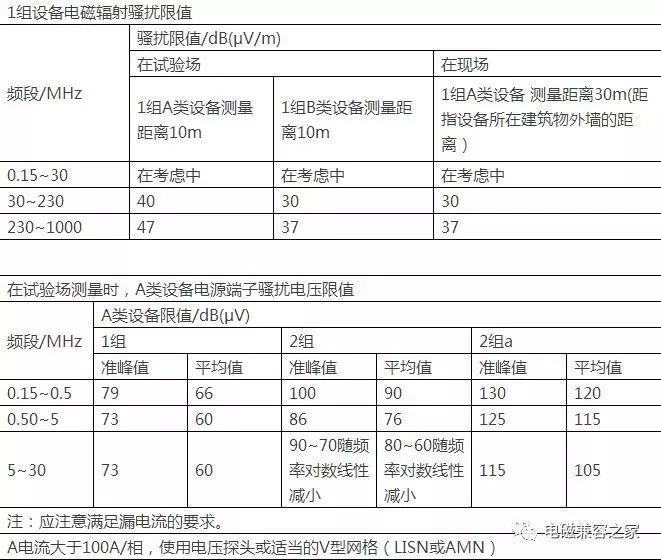


下文为几年前国家食品药品监督管理局关于医疗器械的电磁兼容问题下发的文件:
YY 0505医用电气电磁兼容标准作为与GB 9706.1医用电气安全标准并列的医用电气设备通用安全标准,对于控制产品电磁兼容指标,保证人民群众用械安全起着重要的作用。按照有关标准制修订计划,国家食品药品监督管理局修订完成了YY 0505-2012《医用电气设备 第1-2部分:安全通用要求并列标准:电磁兼容 要求和试验》标准(以下简称电磁兼容标准),代替原YY 0505-2005标准。为保证标准顺利有序实施,现将相关工作要求通知如下:
一、医疗器械生产企业作为保证产品质量的第一责任人,应积极主动学习电磁兼容标准,充分理解并掌握标准要求,充分利用各种社会检测资源,从产品研制阶段开始,做好相应标准实施准备工作。
在电磁兼容标准发布实施后,医疗器械生产企业应立即在研制、生产等全过程中贯彻实施电磁兼容标准,并按照《关于印发进一步加强和规范医疗器械注册管理暂行规定的通知》(国食药监械〔2008〕409号)要求组织生产,确保产品符合电磁兼容标准要求。
二、自电磁兼容标准实施之日起,首次申报注册的第Ⅲ类医用电气设备在注册申报时应提交由医疗器械检测机构出具的符合电磁兼容标准要求的检测报告。在此之前申请注册并获得受理的和已获准注册的第Ⅲ类医用电气设备,在重新注册时再提交符合电磁兼容标准要求的相应检测报告。
自电磁兼容标准实施一年后,首次申报注册的第Ⅱ类医用电气设备,在注册申报时应提交由医疗器械检测机构出具的符合电磁兼容标准要求的检测报告。首次申报注册的第I类医用电气设备提交包含电磁兼容标准要求的全性能检测报告。在此之前申请注册并获得受理和已获准注册的第I、Ⅱ类医用电气设备,在重新注册时再提交符合电磁兼容标准要求的相应检测报告。
三、医用电气设备在实施GB 9706.1标准全项检测时,应对电磁兼容性能按照电磁兼容标准要求实施检测,并对涉及电磁兼容性能的检测出具相应格式要求的检测报告。(检测报告格式另文公布)
如具有GB 9706.1标准承检资格的医疗器械检测机构不具备电磁兼容标准承检资格,可根据本单位工作程序实施分包检测,具备电磁兼容标准承检能力的医疗器械检测机构应积极承接有关分包检测。分包方应对GB 9706.1和由被分包方出具的电磁兼容标准检测报告负责,并按本单位工作程序审核后出具相应检测报告。
生产企业应确保实施GB 9706.1标准和电磁兼容标准检测的产品一致,并与相关医疗器械检测机构及时沟通,通报问题,协助做好有关检测工作。不同医疗器械检测机构之间也应充分做好协调和沟通,监督企业做好有关工作。
对于检测过程中发现的重大问题,如基本性能判据、型号覆盖等问题,应在检测报告备注中详细载明有关问题并注明自身意见,以供具体技术审查部门参考。
四、各级食品药品监督管理部门应督促企业按照法规要求组织生产,并在注册审查过程中严格审查是否符合电磁兼容标准要求。
五、各具备电磁兼容标准承检能力的医疗器械检测机构应充分预估检测量,加大电磁兼容检测实验室建设,加强内部管理,与企业充分沟通,合理选择检测方法,提高检测效率,确保注册检测项目和其他医疗器械检测机构分包检测项目在合同约定时限内完成。
国家食品药品监督管理局鼓励各医疗器械检测机构积极与外系统相应优质电磁兼容实验室资源开展合作共建,并将对有关电磁兼容检测项目认证申请予以尽快处理。
六、在电磁兼容标准实施过程中,如出现不同医疗器械检测机构或技术审评部门等单位对标准条款、要求理解不同等影响标准全面实施的整体性、共性问题,相关单位可向国家食品药品监督管理局医疗器械标准管理中心提出申请,由其根据情况,决定是否启动专家咨询机制,并按程序经专家咨询提出解决建议,报国家食品药品监督管理局医疗器械监管司。
七、检验诊断类医用电气设备执行GB/T 18268.1:2010《测量、控制和实验室用的电气设备 电磁兼容性要求 第一部分:通用要求》标准,可参照本实施通知执行。
各级食品药品监督管理部门应加强协调,保证有关工作的落实,确保标准得以顺利实施。


