基于vasp计算材料红外与Raman光谱信息

使用方法一:获取材料raman活性信息
代码链接:https://github.com/raman-sc/VASP/tree/master/Sibulk-VASP
前置计算材料的振动频率和介电常数等,参考INCAR如下:
SYSTEM = Si_bulk
ISTART = 0 # From-scratch; job : 0-new 1-cont 2-samecut
NWRITE = 3 Verbosity
! electronic relaxation
ENCUT = 300.0 # cut-off energy
PREC = Accurate # precision : accurate/normal/low
ISPIN = 1 # 1 - off, 2 - on (non spin-polarized calculation)
ICHARG = 2 # > 10 for non-SC calculation
IALGO = 38 # DAVidson, then RMM-DIIS
EDIFF = 1.0E-8 # default
ISMEAR = 0 # gaussian
SIGMA = 0.05
! PAW's
LREAL = .FALSE. # default - Automatic choice of how projection is done
ADDGRID = .TRUE.
! phonons
IBRION = 5
POTIM = 0.01
! parallelisation
LPLANE = .FALSE.
KPAR=8
! output
LWAVE = .FALSE. # WAVECAR file
LCHARG = .FALSE. # CHCAR file
LELF = .FALSE.
LVTOT = .FALSE.
将计算得到的OUTCAR和使用的POSCAR后缀加上.phon后,执行运行脚本raman.sub,其中同时会运行vasp_raman.py脚本。具体功能可参考说明。
使用前需根据材料结构对称性和Wyckoff 点位修改脚本参数。
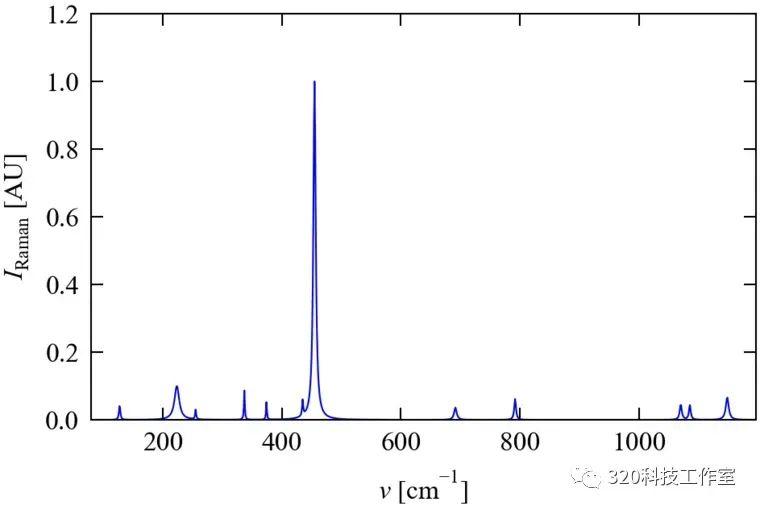
计算得到的硅的Raman活性信息如下。
# mode freq(cm-1) alpha beta2 activity
1 504.47552 0.0000409 780.7952797 5465.5669580
2 504.47464 0.0045779 779.0264832 5453.1863253
3 504.47201 -0.0031882 779.4324411 5456.0275454
4 447.84208 0.0006131 0.0005437 0.0038231
5 447.84107 -0.0017167 0.0000277 0.0003268
6 447.84076 0.0049049 0.0000643 0.0015326
7 447.83928 0.0004905 0.0001417 0.0010030
8 447.83865 -0.0380539 0.0203246 0.2074370
9 447.82907 -0.0295112 0.0906046 0.6734232
10 402.16258 -0.0002044 0.0001276 0.0008950
11 402.16054 0.0001226 0.0000161 0.0001131
12 402.15924 -0.0008992 0.0000119 0.0001199
13 402.15921 -0.0012262 0.0000106 0.0001422
14 402.15746 0.0008584 0.0002334 0.0016672
15 402.15492 0.0005722 0.0000023 0.0000306
16 145.82303 0.0002044 0.0000004 0.0000045
17 145.82113 -0.0000817 0.0000001 0.0000010
18 145.81855 -0.0002044 0.0000002 0.0000029
19 145.81681 -0.0001226 0.0000007 0.0000054
20 145.81480 0.0000817 0.0000004 0.0000029
21 145.81429 0.0000409 0.0000002 0.0000011
活性信息与实验结果相近。(J.H. Parker, et al., Phys Rev, 155, 712 (1967))
使用方法二:Phonopy-Spectroscopy计算材料红外和Raman图像
前置计算:
1、需要通过有限位移法或密度泛函微扰论(DFPT)计算得到材料二阶力常数(有限位移法获得的为FORCE SETS,需通过hiphive或phonopy转化为FORCE_CONSTANTS)。同时可将其转化为hdf5文件。
2、需要计算得到材料的BORN电荷,有限位移法和额外进行一次自洽计算获得,DFPT可一次计算得到。
INCAR参数:LEPSILON = True
3、通过phono3py计算得到材料的三阶力常数,计算任务数量可通过设置位移大小适配计算资源。同时可将其转化为hdf5文件。
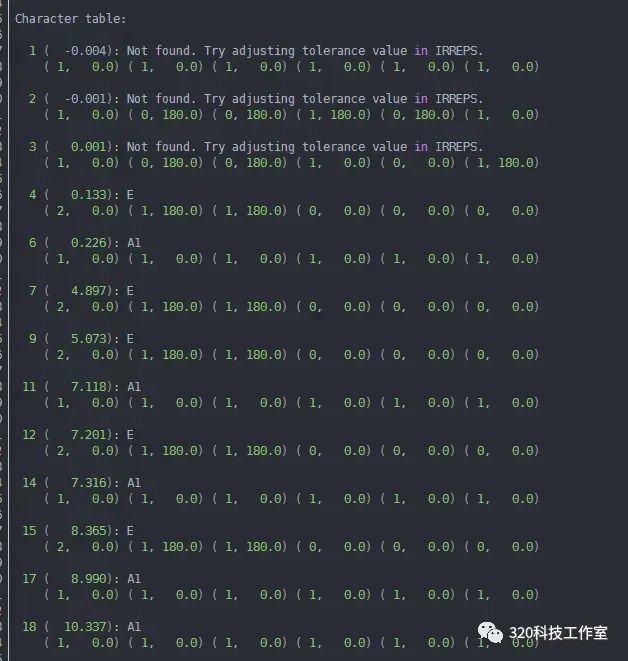
依据前面计算,得到材料在Γ点的振动模式信息,包括mesh.hdf5或mesh.yaml文件和irreps.yaml文件。
获得mesh.hdf5文件:
phonopy --dim="3 3 1" -c POSCAR-unitcell --readfc --hdf5 --fc-symmetry --mesh="1 1 1" –eigenvectors
获得irreps.yaml文件:
phonopy --dim="3 3 1" -c POSCAR-unitcell --readfc --hdf5 --fc-symmetry --irreps="0 0 0"
可根据材料晶体结构以及对称性判断Raman活性信息,也可根据irreps.yaml文件判断
使用Phonopy-Spectroscopy 软件分析前置计算得到的文件并计算红外和Raman图像
生成&Gamma点模式的声子线宽:
phono3py --dim="2 2 2" --dim_fc2="6 6 3" --fc2 --fc3 -v --br --thm --mesh="48 48 48" --write_gamma --gp=0
红外强度计算
这时需要使用之前准备好的BORN文件,生成室温(300 K)线宽的模拟红外光谱和峰值表,命令如下
phonopy-ir --ir_reps --linewidth_hdf5="kappa-m484848-g0.hdf5" --linewidth_temperature=300
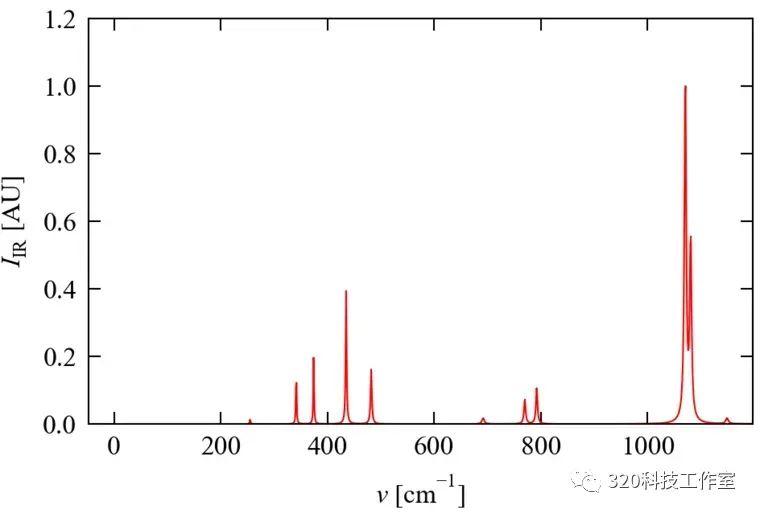
Raman计算
通过irreps.yaml的振动信息,并参考材料结构信息,判断可能显示Raman活动的模式,并生成计算文件:
phonopy-raman -d --bands="4 5 6 7 8 9 11 12 13 14 15 17 18 20 21 22 23 25 26 27"
bands的值为需要计算的模式。
计算的INCAR 参考软件的example,如下
ALGO = Normal
EDIFF = 1E-8
ENCUT = 700
ISIF = 2
ISMEAR = 0
LASPH = .TRUE.
LCHARG = .FALSE.
LEPSILON = .TRUE.
LREAL = .FALSE.
LWAVE = .FALSE.
NSW = 0
PREC = Accurate
SIGMA = 0.01
SYSTEM = SiO2
计算完成后处理OUTCAR的文件
phonopy-raman -r OUTCAR.*
最后获得Raman计算结果
phonopy-raman -p --ir-reps --linewidth-hdf5="kappa-m484848-g0.hdf5" --linewidth-temperature=300