Gromacs分析处理-模拟前后蛋白结构差异对比图的制作

Step1:利用模拟得到的拓扑文件和估计文件,提取模拟前后的两帧结构
1. gmx trjconv -s md_test.tpr -f md_test.xtc -o first.pdb -sep -b 0 -e 0 -pbc mol
#提取首帧
2. gmx trjconv -s md_results_1.tpr -f md_results_1.xtc -o last.pdb -sep -b 100000 -e 100000 -pbc mol
#这里的结构一共100000帧,所以取最后一帧
两者都选择protein进行输出,这样我们就得到了first和last这两个起始结构的pdb结构
Step2:利用pymol预处理
load first0.pdb #加载
load last0.pdb
align last0, first0 #对齐
在命令框输入
Step3:利用pymol脚本(modevectors.py)进行再处理
modevectors.py下载地址:https://pymolwiki.org/index.php/Modevectors
Step4:查看工作路径,也可以更改工作路径,确保你的文件都在此路径下
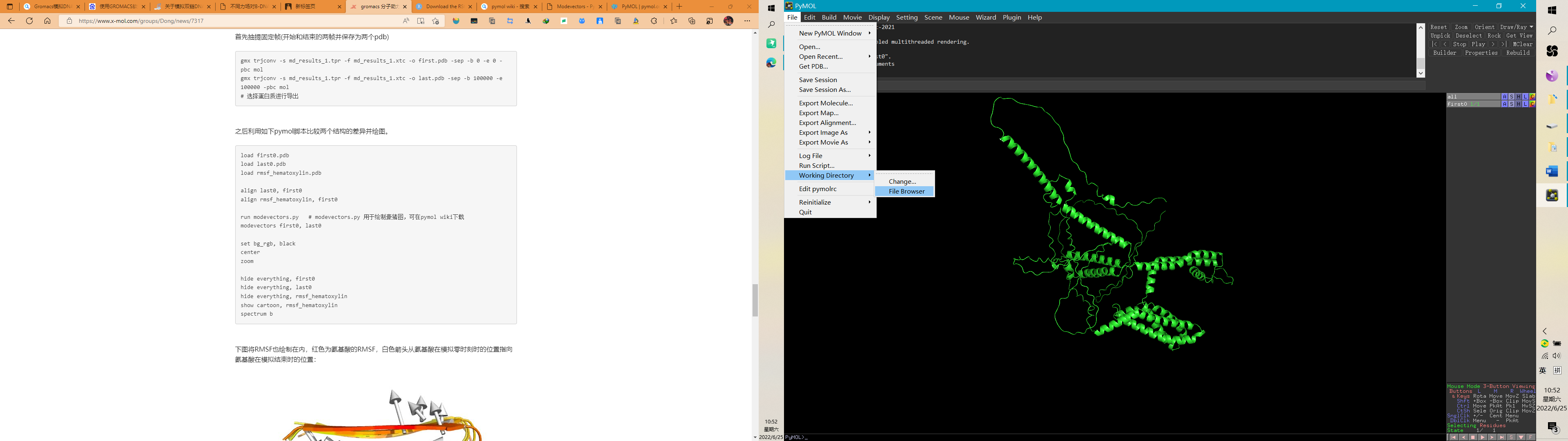

Step5:开始运行pymol脚本,在命令框依次输入
run modevectors.py #用于绘制豪猪图
modevectors first0, last0
center
zoom
show cartoon, first0
show cartoon, last0
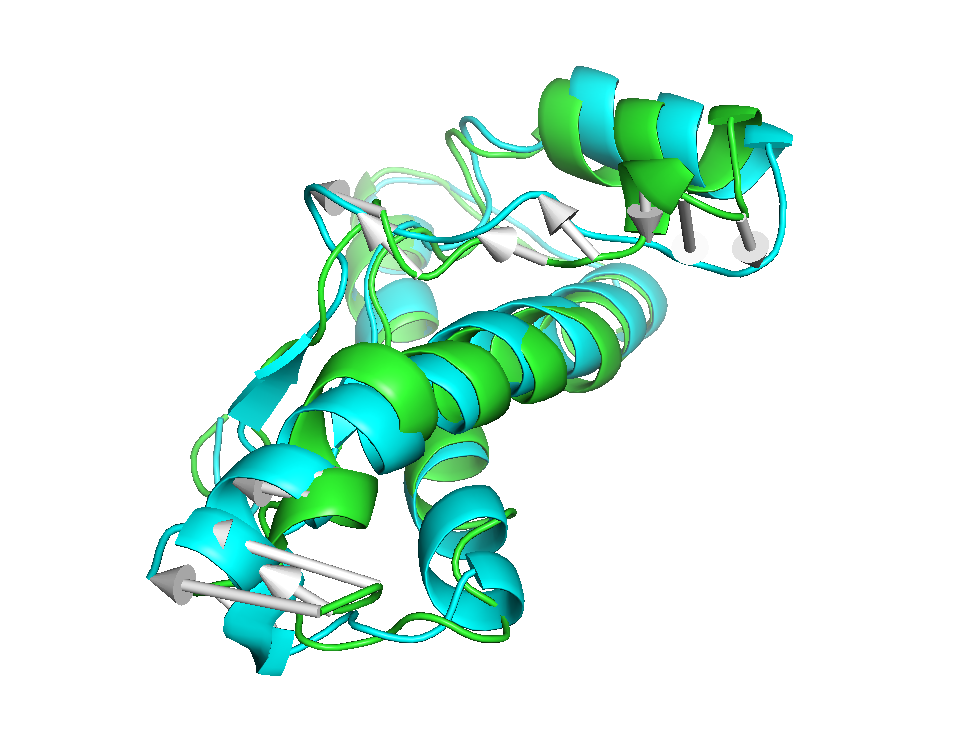
最后的结果,差异体现在箭头处,箭头的指向是从初始到结束位置
其他的颜色,大小,清晰度都可以自己再去调整
最后, 有gromacs相关MD需求,欢迎通过公zhong号联系我们.
公zhong号:320科技工作室
登录后免费查看全文
著作权归作者所有,欢迎分享,未经许可,不得转载
首次发布时间:2023-10-31
最近编辑:1年前

硕士
|
结构工程师
lammps/ms/vasp/

相关推荐
最新文章
热门文章
