Gromacs通过伞状抽样的方法计算自由能

随着计算机技术的发展和计算能力的提升,以模拟和计算的方式去探究实验科学中无法看到的分子机制和细节对目前的科学研究具有极大的促进作用。在分子动力学模拟领域,具有众多成熟的模拟软件例如NAMD、LAMMPS、Amber以及Gromacs等。其中Gromacs不仅对大量的算法进行了优化也更利于初学者上手,例如具有更好的兼容性,更多的分析命令以及操作更加清晰等等。本次简要介绍利用Gromacs的伞状抽样的方法计算结合能。
首先要使用gmx pdb2gmx指令产生对应分子的力场文件和结构文件,并将产生的结构文件放置在盒子中。在该步骤需要注意由于伞状抽样需要用到更大的空间,所以必须要在对应的维度增大盒子尺寸。

随后进行的就是添加水和离子进行能量最小化以及后续较短时间的NVT和NPT模拟。在进行后期的MD模拟时,需要自定义索引组进行牵引,并设置mdp文件中的牵引参数例如速度和力等。

此时模拟过程中的牵引力会被记录在xvg文件中,力随时间变化曲线如图所示:
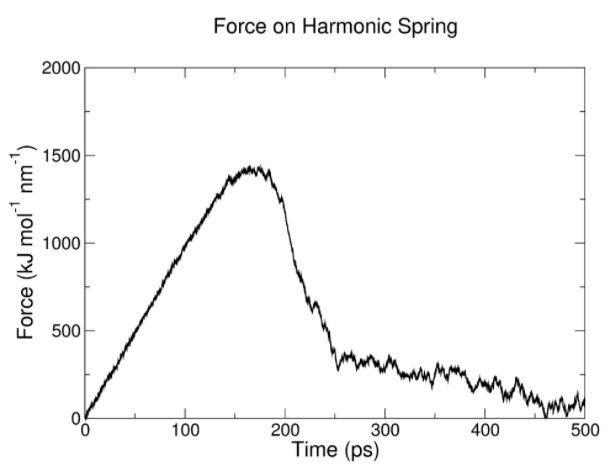
在该过程中,我们可以设定模拟轨迹产生的帧数,然后通过得到单链拉离的多个轨迹分别进行多组模拟。通过在模拟中设定牵引力,将其限制在对应选定的窗口中,即可得到多组模拟产生的pullf.xvg文件。
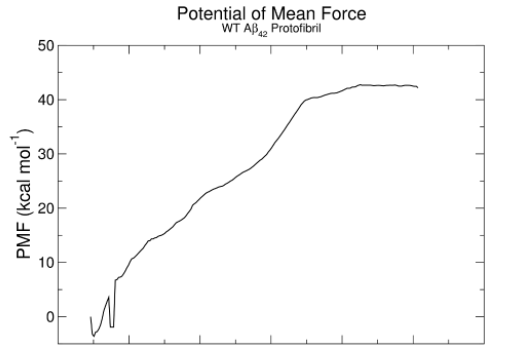
自由能通常可从伞状抽样获取的PMF得到即PMF曲线最高值和最低值的差值。通过使用gmx wham指令打开每一个模拟的tpr文件和pullf.xvg文件,就可以得出PMF值随距离的变化图,如下图所示:
结语
本文简要介绍了Gromacs中通过伞状抽样计算结合能的流程。在实际的操作过程中,用户需要准确了解每个命令中的用途以及如何针对自己的需要选择合适的mdp文件中设定例如计算时长,输出频率,牵引参数等。
最后,有相关需求,欢迎通过公棕号联系我们
公棕号:320科技工作室。


