lammps在金属位错动力学上的应用
本文摘要(由AI生成):
本文介绍了位错动力学在材料科学研究中的重要性,以及利用分子动力学方法模拟单一位错运动的研究案例。文章详细描述了模拟的初始模型、模拟过程以及位错提取的方法。模拟结果显示,不同合金含量的单一位错开动情况不同,位错在克服晶格阻力后会发生位移突变。本文的研究有助于更深入地理解位错运动的规律,为材料性能的改进提供理论依据。
1、位错动力学计算模拟介绍
位错在材料科学 中,指晶体材料 的一种内部微观缺陷,即原子的局部不规则排列(晶体学缺陷)。从几何角度看,位错属于一种线缺陷 ,可视为晶体中已滑移 部分与未滑移部分的分界线,其存在对材料的物理性能,尤其是力学性能,具有极大的影响。位错动力学在材料行为的介观机制研究具有重要的意义。


金属材料在单向拉伸时,由弹性阶段转为塑性阶段,其内在机理为晶格畸变不能满足原子稳定排列存在,产生位错滑移,位错的运动可以体现原子差排组织的运动。研究位错运动规律,就是在研究晶体内原子组的运动规律。研究单个位错的运动问题,可以更准确清晰地理解认知位错运动的规律,从而得出定量的计算结果。对于研究工具,主要是分子动力学。分子动力学计算是利用原子间的势函数关系,对每个原子的独立运动进行计算,是分子运动计算的基础运算工具,计算结果依靠势函数,准确度高,可靠性大,但是计算量大,时间成本高。分子动力学适合对少量位错的运动进行精准计算。本次我们讨论研究的是单独位错运动的定量计算,所以适合运动分子动力学的方法进行。
2、模拟案例
初始模型
比如研究Fe-Cr里单一位错的运动机制。使用atomsk建造一个单一位错,将建造的数据文件导入lammps中,因为位错容易在滑移面发生滑移,所以对于bcc结构的铁素体首要模拟的是材料在110面向111方向滑移。
模拟过程
在弛豫、温度0K条件下,对刃位错模型沿x轴,对螺位错模型沿z轴施加一个持续不变的切应力,由(文献)可知位错的派纳力在150Mpa以内,所以每组不同合金元素的模型,从0~150Mpa做151组不同切应力下位错运动计算。
提取位错
由于派-纳力是位错克服晶体点阵阻力的最小开动力,位错产生瞬间移动其实是微观原子发生了晶体剧烈振动,所以位错开动的位移距离其实是非常小的,大约在几个A左右,所以必须要对位错进行提取,才能准确地定位位错的位置。
在lammps中位错线的可视化提取可以观测位错的移动情况,但是无法得到文本数据,所以需要凭借编程程序进行提取。主要使用的是Julia对文本数据进行处理。而提取的主要依据就是晶格畸变参数,位错的位置就是晶体晶格畸变参数明显的原子组的定位,在lammps模拟过程中,我们添加了晶格畸变参数的计算,以便对数据文本的处理。提取后的效果是比较明显的,模型中最后只留下了产生位错的原子组合。

位错提取前(左)与提取后(右)
模拟结果
由于合金含量不同,单一位错开动的是不一样的。如下图所示,单一位错在持续恒力的条件下,当持续应力达到一定值时,位错会发生一个明显的运动,此时我们可以判定位错克服晶格阻力产生了位移突变,即克服能垒进入热驱动状态,而随着应力的增大,位错的运动并不能很明显地产生,这是由于应力增大后位错开动后所剩的力会导致位错周围的原子再次对位错产生反作用或者钉扎从而使位错发生反向运动或者依然保持不动。

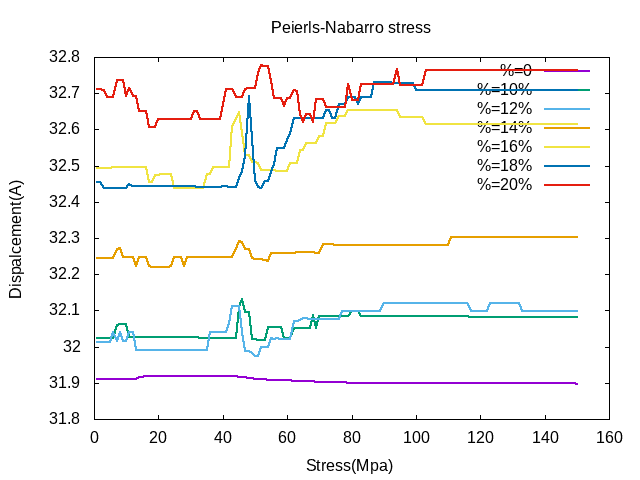
刃位错(左)和螺位错(右)不同Cr比例下的P-N力
最后,欢迎大家联系我们。
公棕号:320科技工作室。



