量子计算(Quantum computing)在工程应用和力学当中的应用研究
文一:

量子计算增强的距离最小化数据驱动的计算力学
摘要:
通过消除材料建模误差和不确定性,距离最小化数据驱动计算力学在工程应用中具有巨大潜力。在这个计算框架中,求解过程依赖于最小化本构数据库与守恒律之间的距离。然而,在大型数据库的情况下,距离计算是非常耗时的,往往占用了大部分的计算时间。在这篇文章中,我们展示了如何使用量子计算来增强数据驱动的计算力学,以指数方式降低距离计算的计算复杂度。该方法不仅在量子计算机模拟器 Qiskit 上得到了验证,而且在 OriginQ 的实际量子计算机上也得到了验证。我们相信,这项工作代表着在将量子计算融入数据驱动的计算力学方面迈出了充满希望的一步。

图:互换测试的量子电路。

图:自适应策略的说明。(a) 由于距离估计中的误差,找到正确的最近数据是一项挑战。(b) 然而,数据的翻译减少了误差,增加了找到正确的最近数据的概率。

图:QDD的计算框架

图:OriginQ量子计算机上用于距离估计的量子电路。

图:模拟器和 OriginQ 量子计算机上 qDD 的演化曲线。
文二:

固体力学与结构工程的量子计算——用变分量子特征解算器进行演示
摘要:
变分量子算法利用叠加和纠缠的特性,通过操纵量子态来有效地优化成本函数。它们适用于噪声中等规模量子(NISQ)计算机,这些计算机最近被世界研究界所使用。在这里,我们在IBM Qiskit Runtime平台上实现并演示了5量子位和7量子位量子处理器上的数值过程。我们将商业有限元法(FEM)软件abaqus与变分量子本征求解器(VQE)的实现相结合,建立了一个集成的管道。使用三个实例来研究性能:六边形特拉斯、Timoshenko梁和行星-大气连续体。我们使用这种混合量子经典方法对基频估计的收敛性进行了参数研究。在不久的将来,当具有数百个量子位的量子计算机可用时,我们的发现可以扩展到具有更多自由度的问题。

图:力学问题的变分量子特征解算器算法的流水线。蓝色的步骤表示abaqus预处理,绿色的步骤表示VQE实现,剩下的黄色步骤表示迭代收敛。左侧虚线框插图中显示了一个示例刚度矩阵。“硬件有效模拟”[23,24]的一个特定实例如中心虚线框插图所示,其中以具有6个CX门和深度=1的纠缠模式为例。补充材料[25]中提供了其他示例。右侧虚线框插图列出了用户定义的关键参数。

图:三个不同案例研究的示意图: (a)六边形桁架,(b) Timoshenko 梁,和(c)平面应变连续体。黄点/黄线所示位置订明边界条件。

图:VQE结果的错误,如等式(12)所定义的,对于情况(i):(a)具有纠缠模式CZ和深度3的不同优化器选择。3.(c)与优化器不同的纠缠深度Cobyla和纠缠模式CZ。
文三:

走向量子计算力学
摘要:
量子计算的快速发展开创了计算机模拟的新时代,为不同学科提供了突破性的机会。这场革命的核心是量子处理器纠缠量子位的能力,为解决极端规模的计算挑战打开了前所未有的可能性,远远超出了经典计算的范围。在这项研究中,我们探索了如何利用量子计算来增强计算力学。我们的重点是在多尺度固体力学的框架内对代表性体积单元(RVE)进行分析。我们介绍了一种创新的量子算法,旨在解决RVE问题。该算法能够在O(Poly-log(N))时间内计算离散化大小为N的RVE,从而实现了比通常与N成线性比例的传统经典计算方法的指数加速。我们通过案例研究验证了我们的方法,包括一维和二维泊松方程的解,以及具有分段恒定相位的复合杆的RVE。我们提供的量子电路设计只需要O(Poly-log(N))通用量子门,这突出了我们方法的效率。我们的工作提出了一种将量子计算与计算力学相结合并对其产生影响的主要方式。
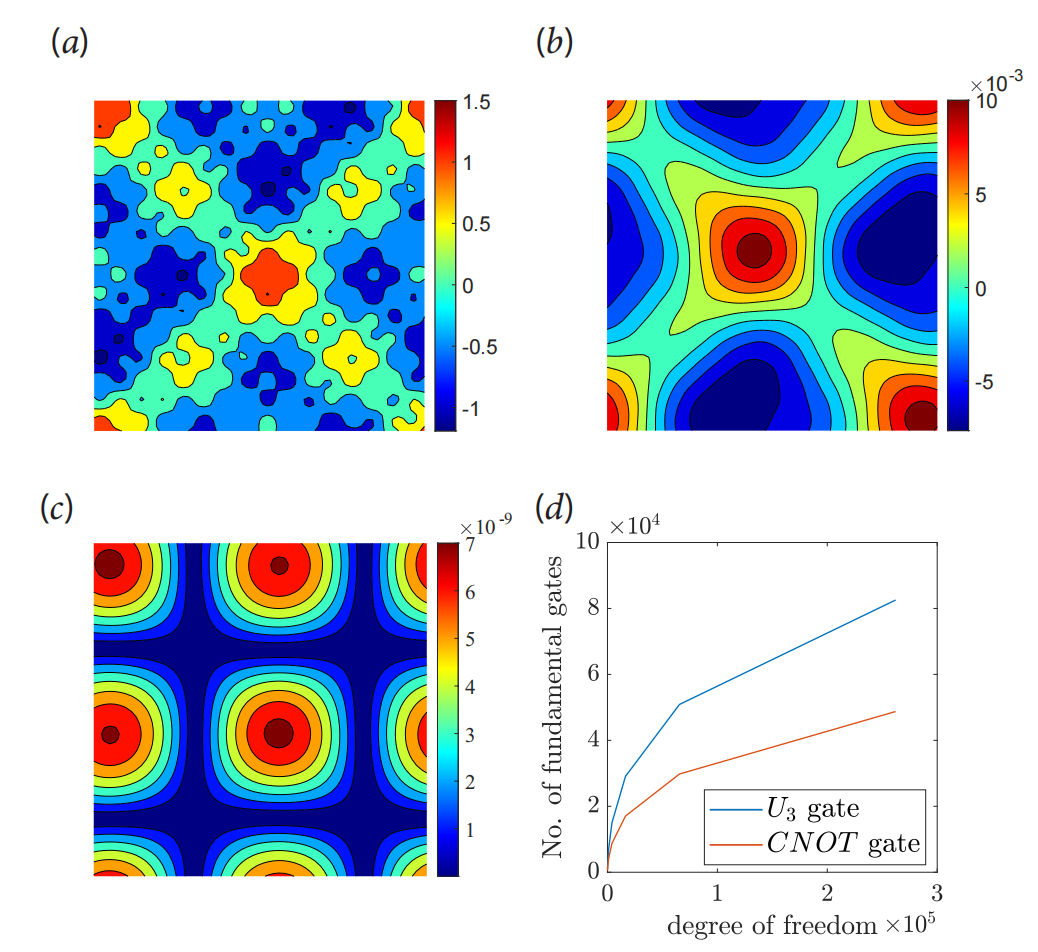
图:二维周期泊松问题。
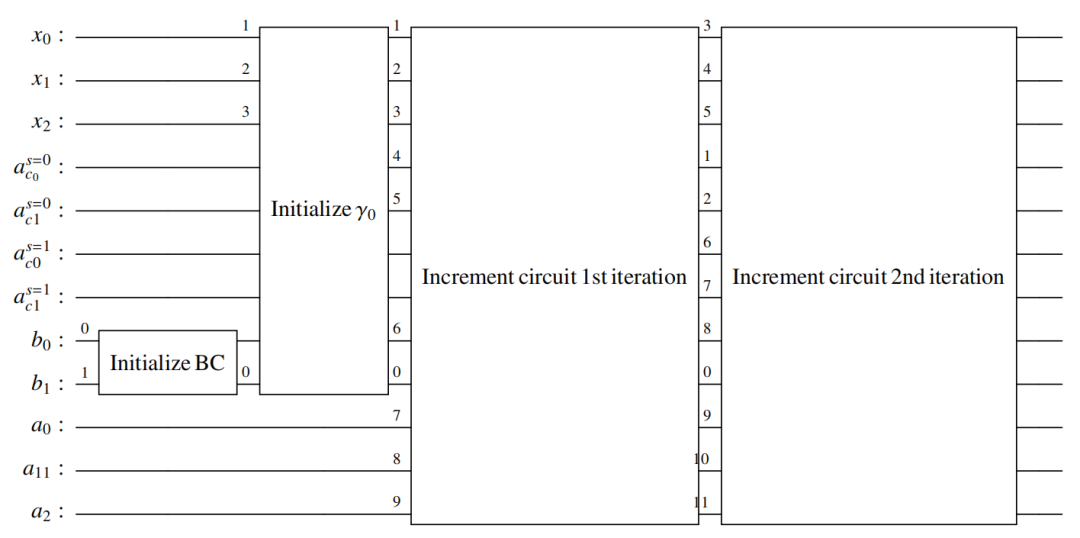
图:两次迭代8自由度 QRVE 算法的定点电路原理图。

图:二维泊松方程的结果。
文四:

量子计算在分子科学中的应用前景
摘要:
分子科学是由电子和原子核的动力学以及它们与电磁场的相互作用所支配的。对这些过程的真实的物理化学理解对于设计和合成对我们的社会和经济有价值的化学品和材料是至关重要的。虽然这个领域的一些问题可以通过经典力学来充分解决,但是许多问题需要明确的量子力学描述。这样的量子问题需要波函数的表示,波函数随系统大小呈指数增长,因此应该自然而然地从一些逻辑量子位的量子计算中受益,这些量子位仅随系统大小线性增长。从这个角度,我们阐述了量子计算在分子科学,即分子物理学,化学,生物化学和材料科学中的潜在好处。
表:QPE和VQE算法的比较

文五:

近期量子计算机上材料的量子模拟
摘要:
量子计算机有望实现对分子和材料特性的有效模拟;然而,由于量子位的数量有限,目前它们只允许对少数原子进行从头计算。为了利用近期量子计算机的力量来模拟更大的系统,需要开发混合量子经典方法,其中量子计算仅限于系统的一小部分。这对于分子和固体特别相关,其中活性区域需要比其环境更高水平的理论精度。在这里,我们提出了一种量子嵌入理论,用于计算有源区的强相关电子态,系统的其余部分在密度泛函理论中描述。我们通过研究半导体中的几个缺陷量子比特来证明该方法的准确性和有效性,这些缺陷量子比特对量子信息技术非常感兴趣。我们在量子计算机上进行计算,并表明它们产生的结果与在经典体系结构上精确对角化获得的结果一致,为在近期量子计算机上模拟真实材料铺平了道路。
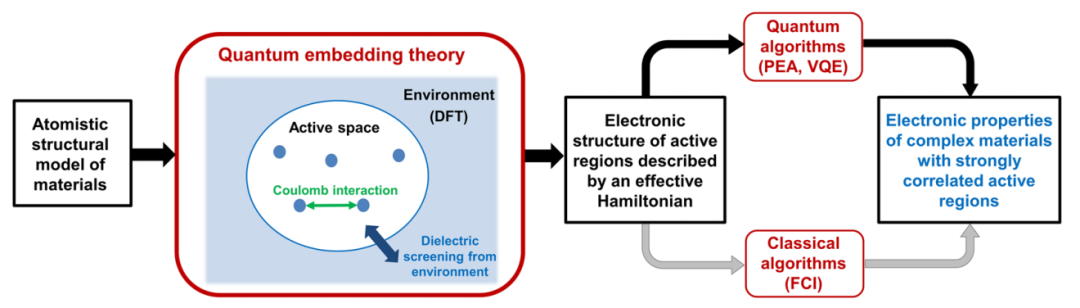
图:使用量子嵌入对材料进行量子模拟的一般策略。整个系统被分为有源空间及其环境,有源空间中的电子态由有效哈密顿量描述,该有效哈密顿量使用经典(例如,全组态相互作用,FCI)或量子算法(例如,相位估计算法(PEA)、变分量子本征解算器(VQE))求解。活性空间中电子之间的有效相互作用包括裸库仑相互作用和由环境的介电屏蔽引起的极化项(见正文),该极化项包括交换相关相互作用进行评估。
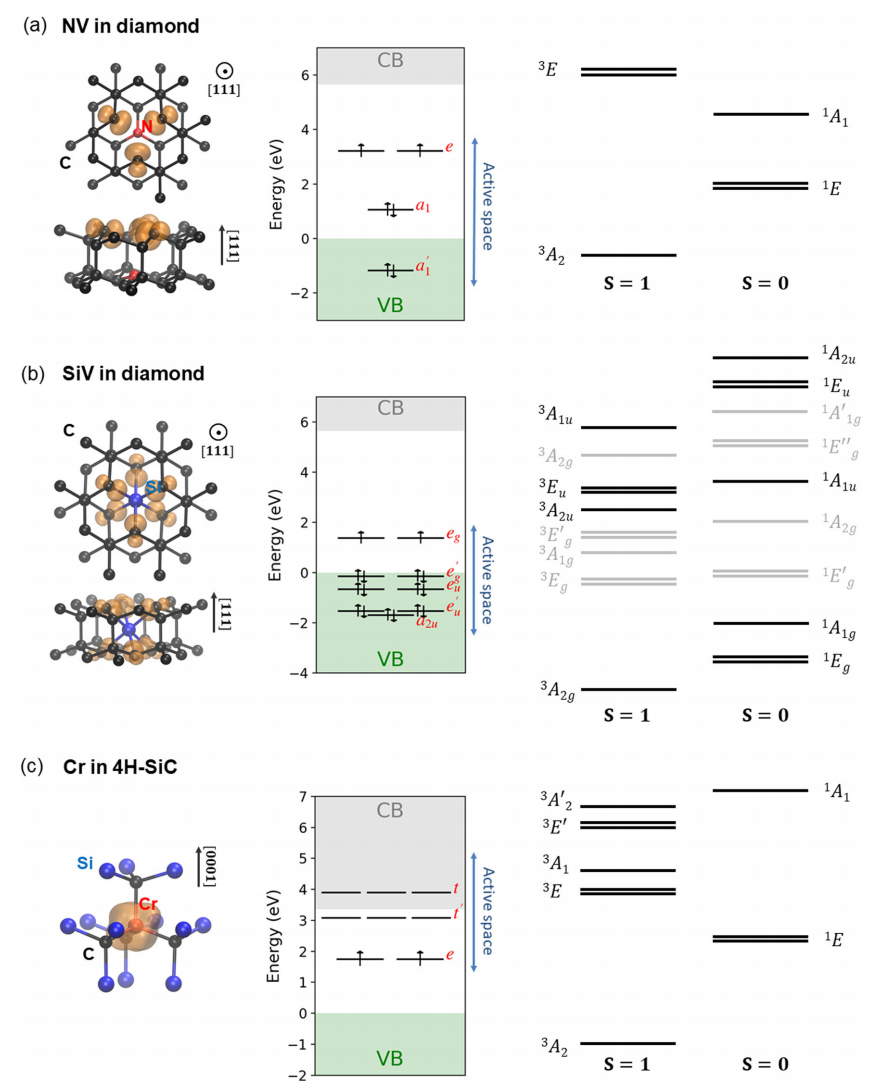
图:自旋缺陷的电子结构。(a) ,(b)和(c)分别给出了金刚石中带负电荷的氮空位(NV)、金刚石中的中性硅空位(SiV)和4H-SiC中的Cr杂质(4+)的结果。左图显示了从自旋不受限制的DFT计算中获得的自旋密度。中间面板显示了通过自旋限制DFT计算计算的单个粒子缺陷水平的位置。活动空间中包括的状态(请参见文本)由蓝色竖线表示。右图显示了通过包括交换相关相互作用的有效哈密顿量的精确对角化(FCI计算)获得的低地多体电子态的对称性和有序性。
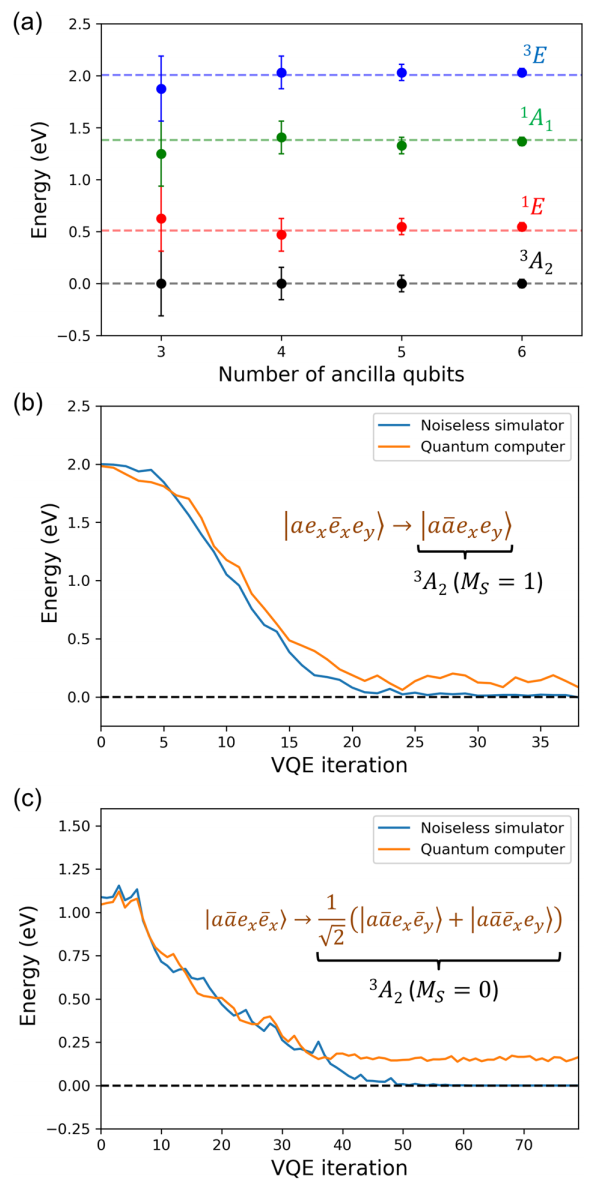
图:利用相位估计算法(PEA)和变分量子特征解算器(VQE)对钻石中 NV 中心的最小模型进行了量子模拟。